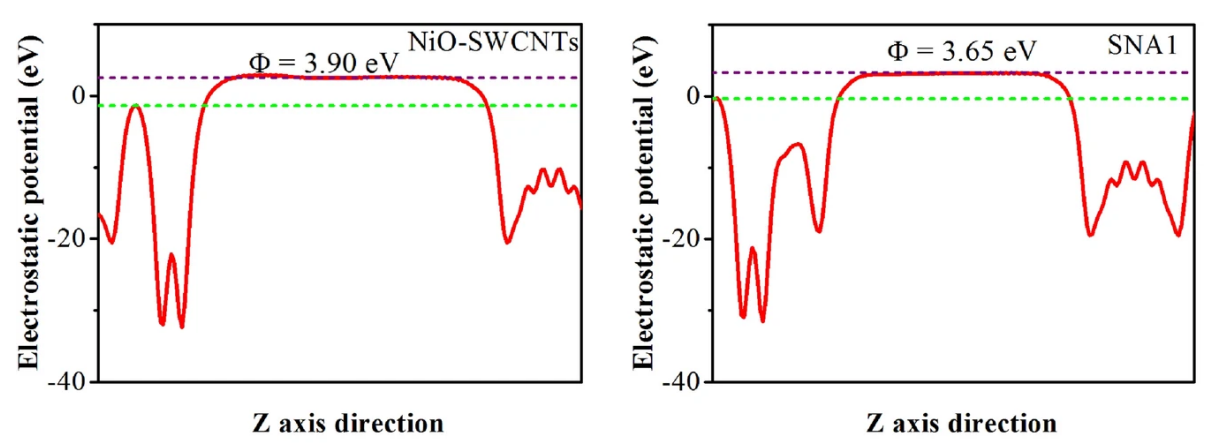
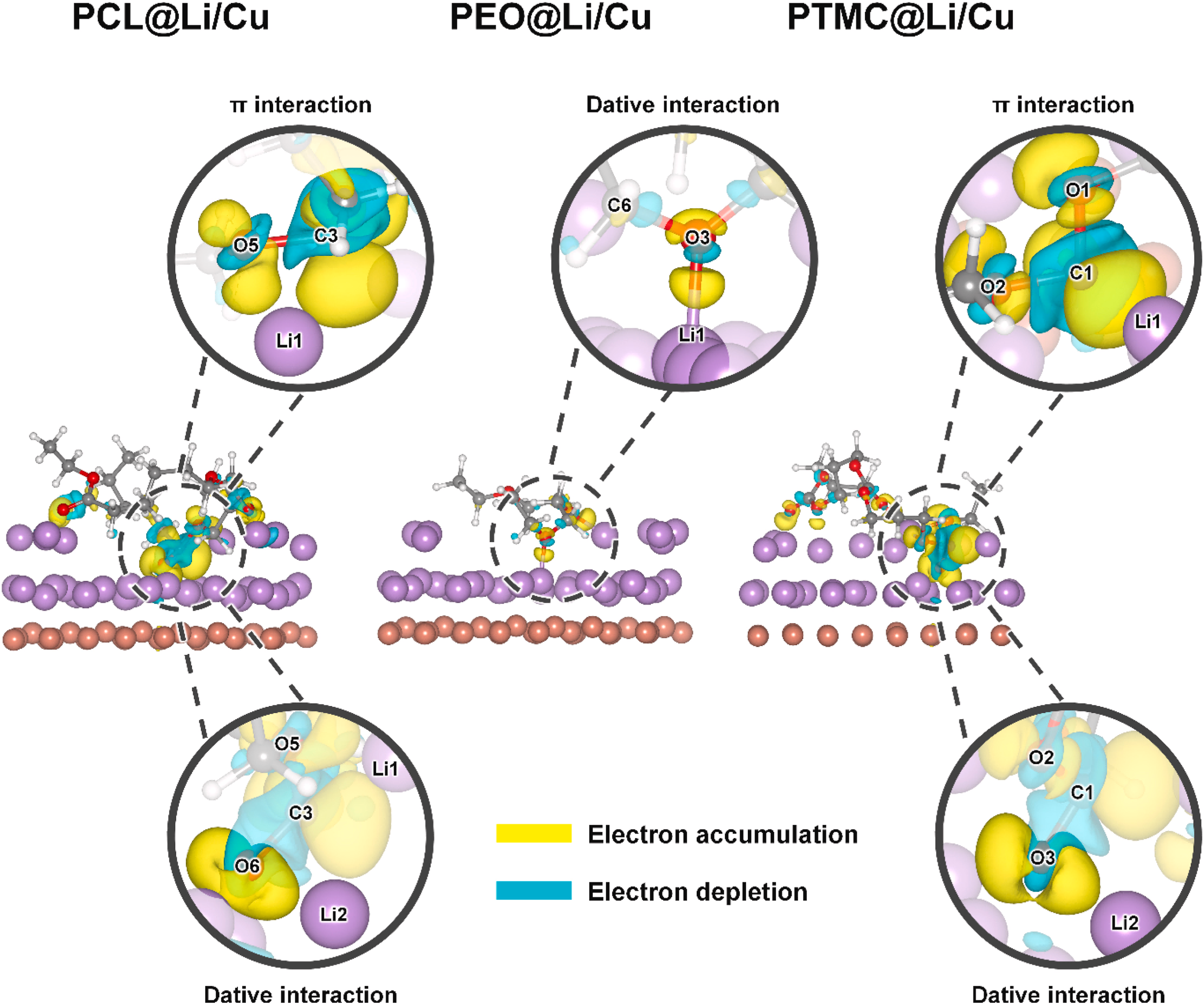
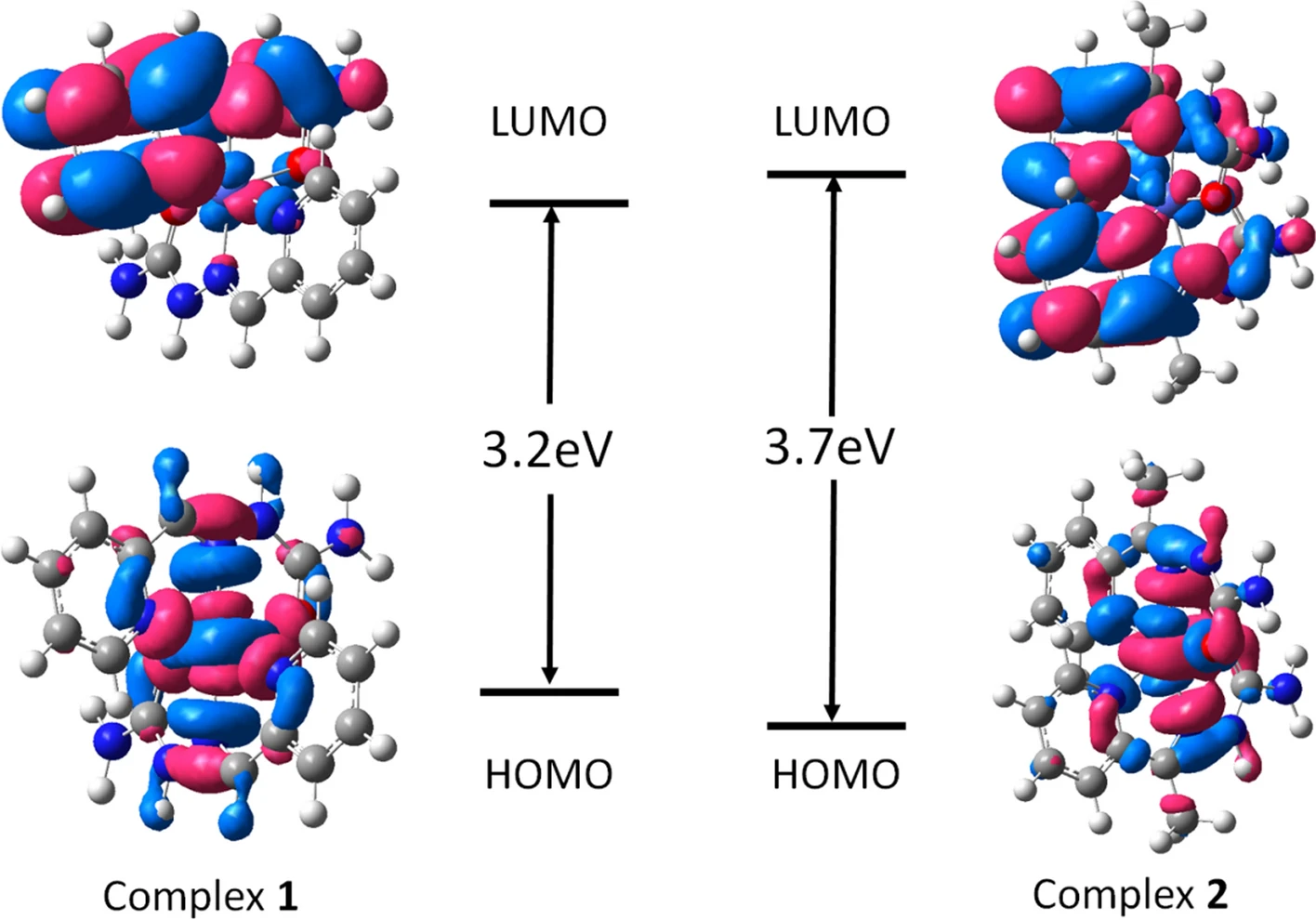
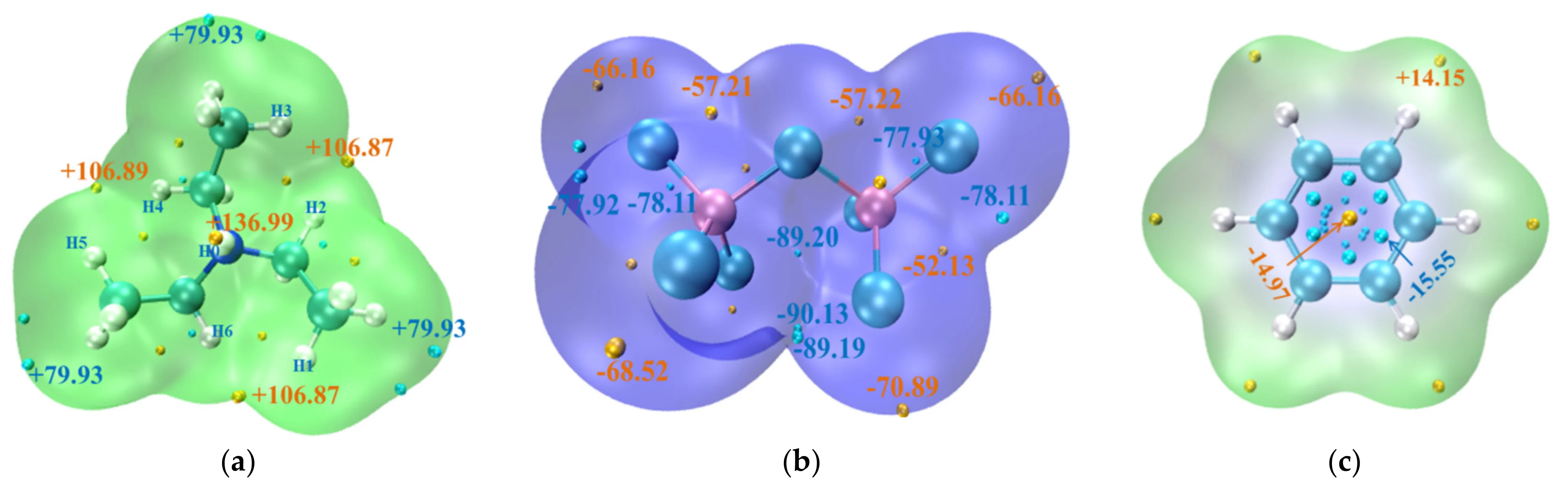
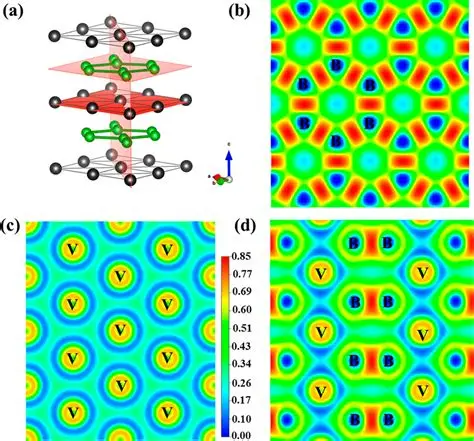
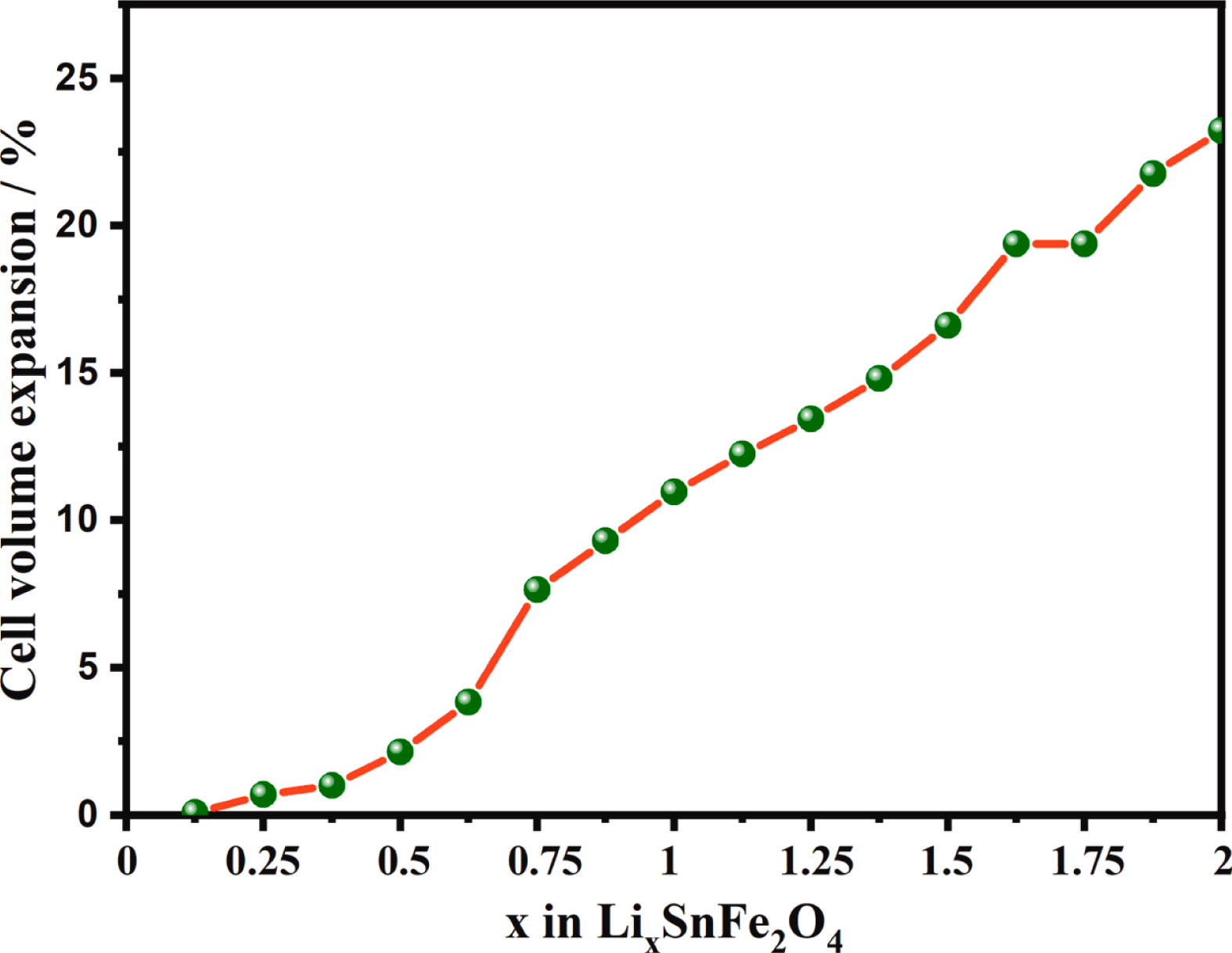
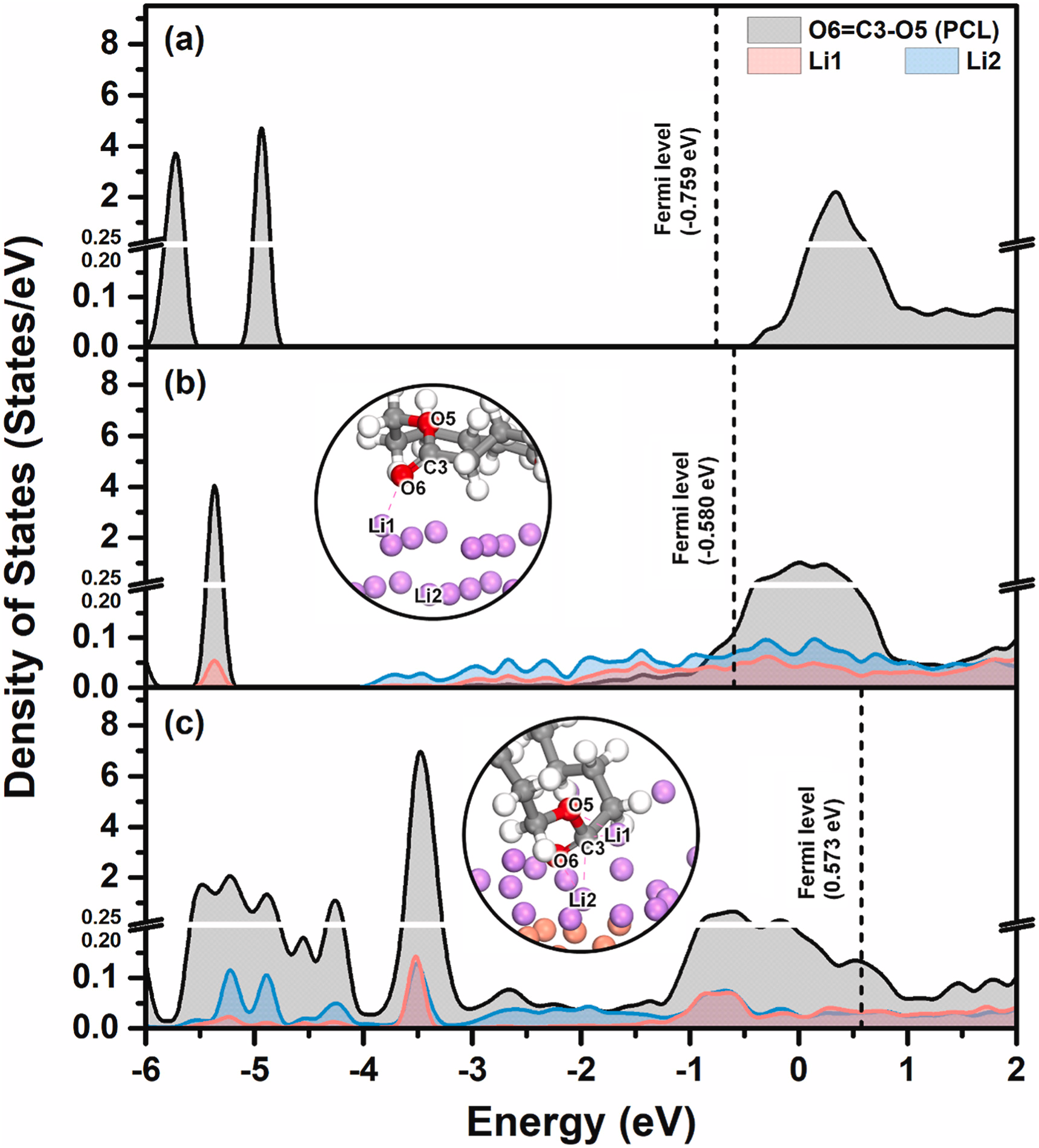
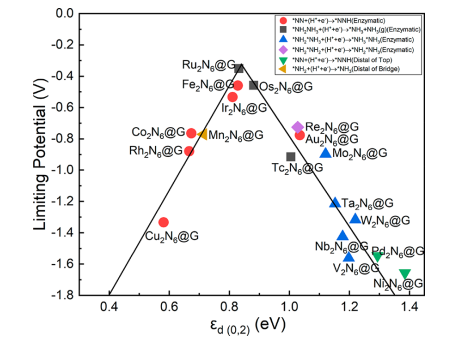
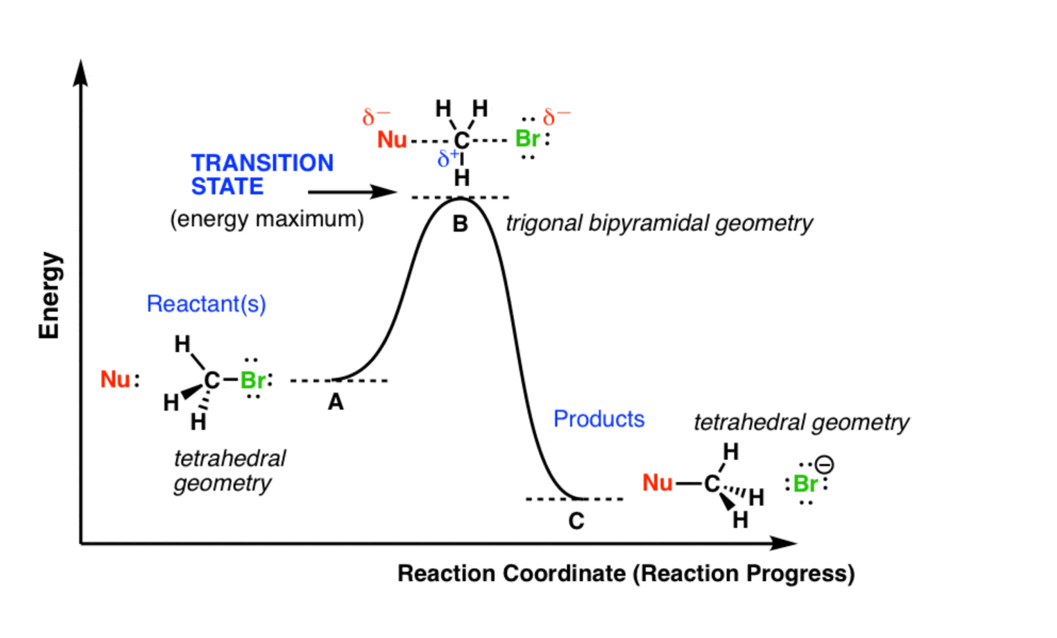
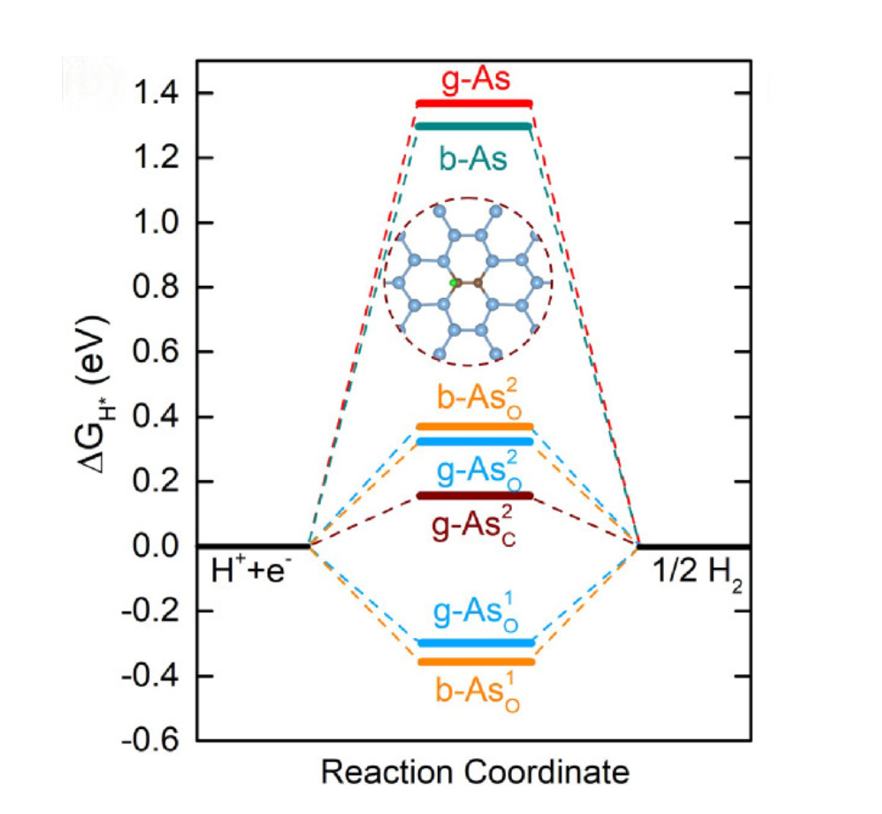
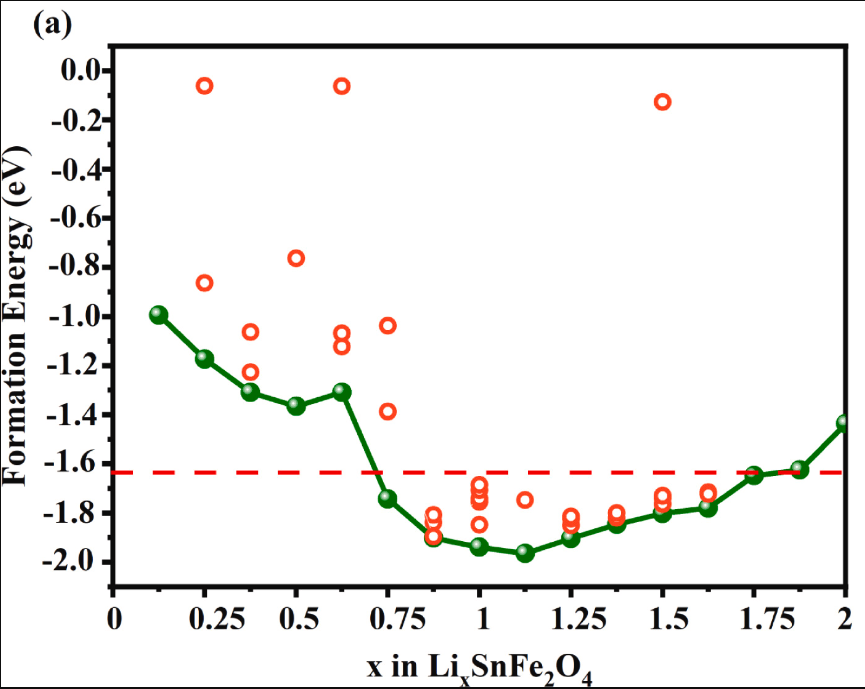
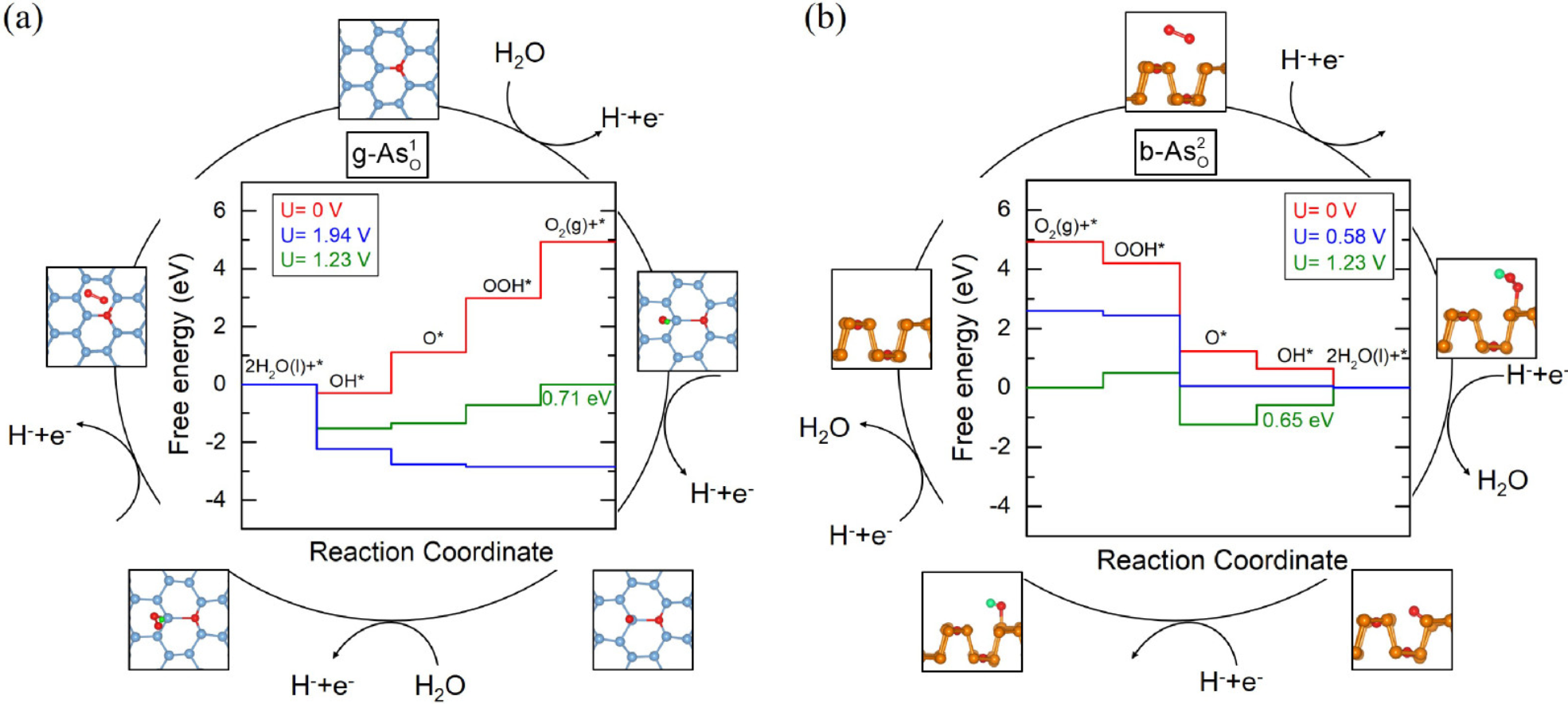
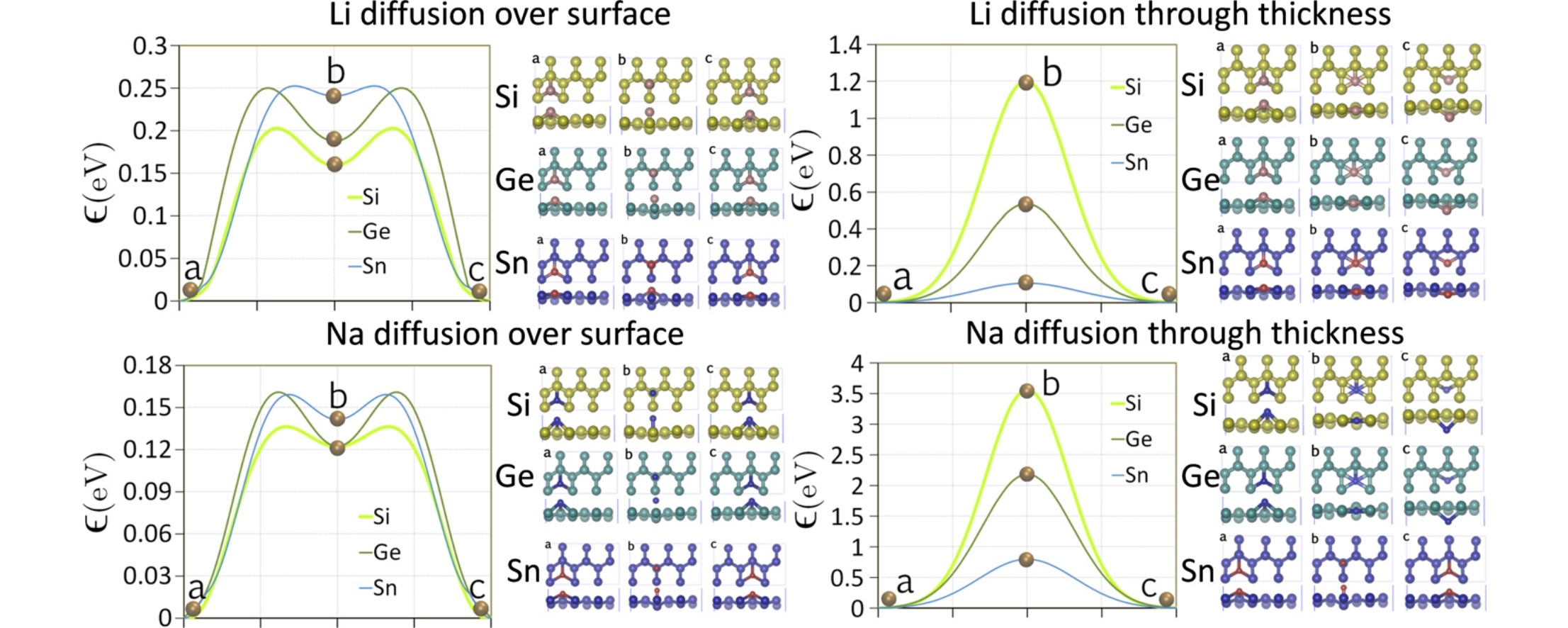
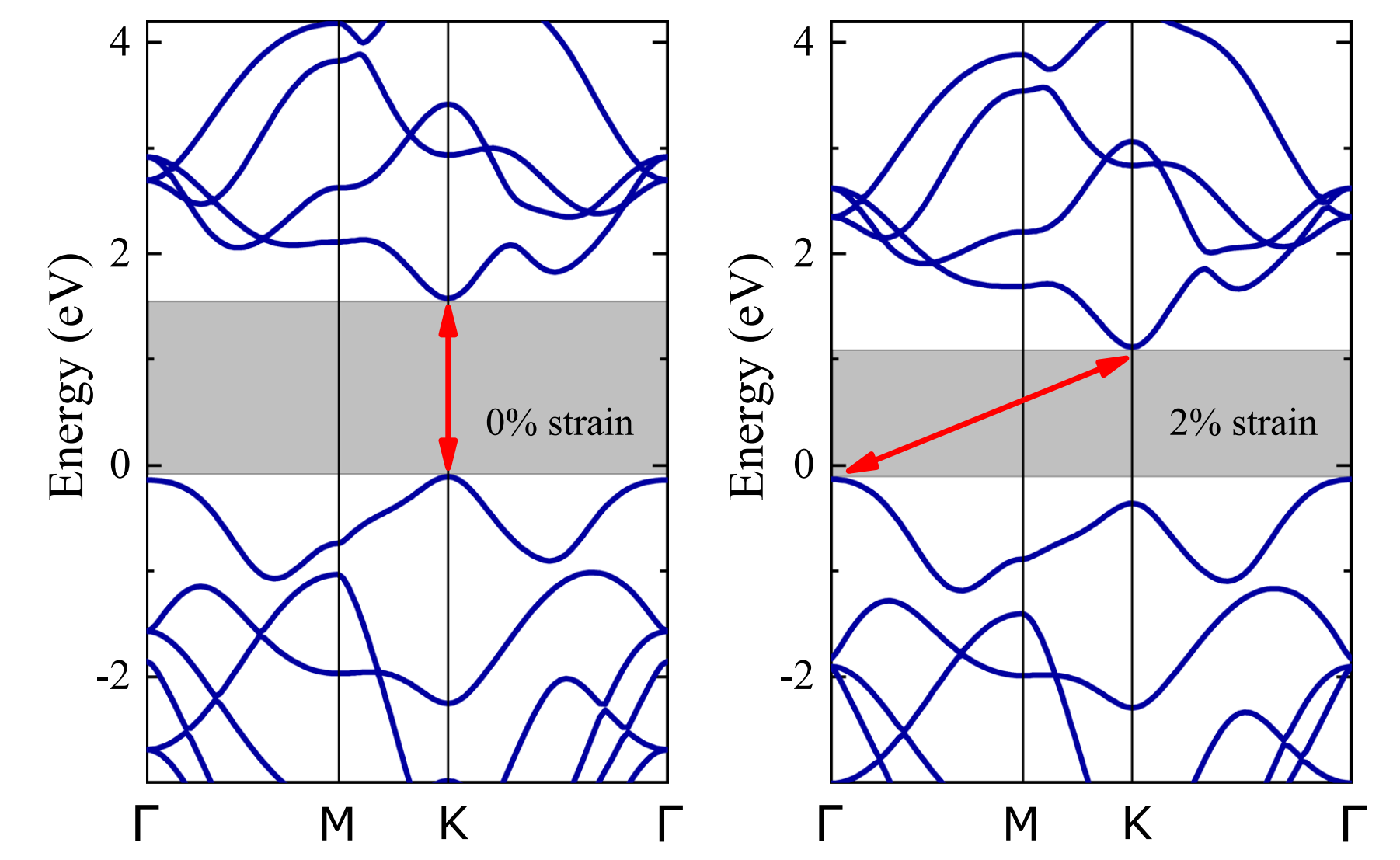
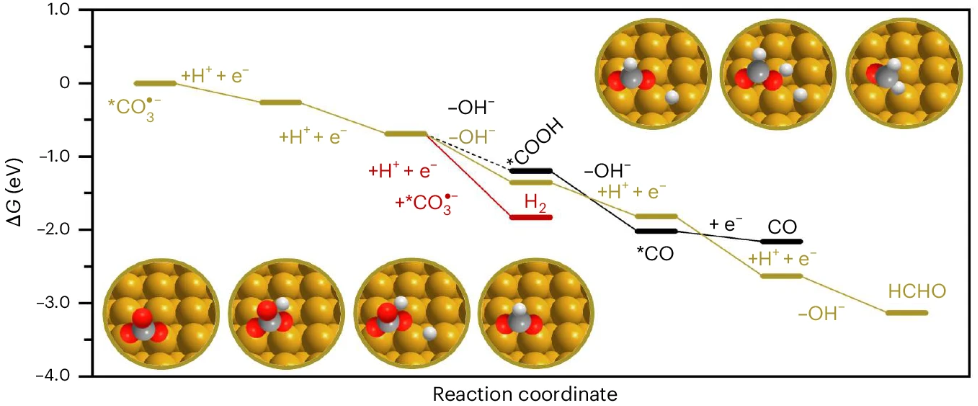
模拟计算
第一性原理计算
基于量子力学、无经验参数的从头算方法,仅依赖基础物理常量,即可精准预测材料的结构、电子性质与反应活性,为实验筛选与机理解析提供核心理论支撑,广泛应用于催化、材料等科研领域。
适用体系金属、合金、氧化物、分子筛、二维材料、钙钛矿、锂电电极材料、催化剂表面与界面、缺陷体系、固溶体等晶体与非晶材料。
可模拟过程吸附、扩散、相变、催化反应路径等微观动力学过程。